Signed in as:
filler@godaddy.com

"...it takes all the running you can do, to keep in the same place"
- the Red Queen in Lewis Carroll's Through the Looking Glass

How do host cells distinguish between pathogens and non-pathogenic microbes? A major focus of the lab is to understand how innate immune sensors detect contextual cues of pathogenic infections, also known as 'Patterns of Pathogenesis.'

Our lab studies a class of innate immune sensors that form inflammasomes, and are particularly interested in how they sense and respond to pathogen-specific activities.

We plan to leverage our understanding of species-specific host-pathogen interactions to generate improved models of human infectious disease.
Host-pathogen interactions are key molecular battlegrounds. These interactions are often zero sum games, where the success of one side comes at the detriment of the other. As a consequence, host-pathogen interactions rapidly evolve in recurrent cycles of adaptation and counter-adaptation. Our lab studies the evolutionary history of host-pathogen 'genetic conflicts' to gain insights into extant host-pathogen interactions that influence human infectious disease.
Want to learn more?
Here's two examples of how molecular evolution can be used to study host-pathogen interactions:
And check out this wonderful review on the subject from our friends Matt Daugherty (UCSD) and Harmit Malik (Fred Hutch).

Rules of engagement: molecular insights from host-virus arms races
by Daugherty & Malik (2012)
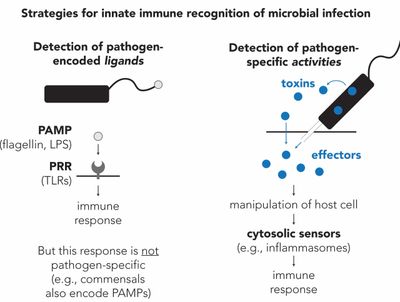
Host cells rely on innate immune sensors called pattern recognition receptors (PRRs) to distinguish between self from non-self (left). These proteins recognize highly essential and conserved molecules that are specific to microbial life, termed pathogen-associated molecular patterns or PAMPs. Years of research has demonstrated the importance of these recognition events for an effective and appropriate immune response.
However, non-harmful, and even beneficial (i.e., commensal) microbes also encode PAMPs. Can the immune system also distinguish between pathogens and non-pathogenic microbes?
We are interested in an alternative mode of innate immune recognition that recognizes pathogen-specific activities. A key feature of pathogens is invasion and manipulation of the host intracellular environment (e.g., immune inhibition, resource acquisition, cellular damage) (right). The activities are essential for the replicative success of pathogens, and thus both widespread and highly conserved. For these reasons, we reason that pathogen-specific activities represent prime targets of innate immune recognition.
The concept of Patterns of Pathogenesis was formalized in this classic Perspective by Russell Vance, Ralph Isberg, and Dan Portnoy.
The MITCHELL LAB has focused on a class of innate immune sensors that detect pathogen-specific activities called inflammasomes. You can read more about that here.

What are inflammasomes?
A major arm of our research focuses on a class of innate immune sensors that form inflammasomes. Inflammasomes are large, cytosolic immune complexes that initiate protective inflammatory responses (e.g., cytokines and pyroptotic cell death). Inflammasomes are critical in host defense against pathogens. However, the inappropriate activation of inflammasomes can cause sever autoinflammatory disease.
How do inflammasomes sense pathogens?
Upon pathogen detection, inflammasome-forming sensors assemble to serve as a platform for the host protease CASP1 (and other inflammatory caspases), which then processes downstream effectors to initiate a proinflammatory response.
Some inflammasomes, like NAIP–NLRC4, detect pathogens by binding to intracellular ligands (e.g., cytosolic DNA, bacterial flagellin). In these cases, ligand binding induces a conformational change in the sensor, which triggers inflammasome assembly. In contrast, other inflammasomes do not detect ligands, and instead detect pathogen-specific activities.
One of these sensors is called NLRP1. In mice, NLRP1B had long been known to be cleaved and activated by the Bacillus anthracis (anthrax) toxin lethal factor (LF) protease. However, the mechanism of how this activity (i.e. a protease toxin) was a) sensed by NLRP1B, and b) linked to inflammasome activation, was unknown. We found that LF-induced proteolysis of the N-terminal domains of NLRP1B leads to their proteasome-mediated degradation. However, because of a self-cleavage event in the NLRP1B C-terminal function-to-find domain (FIIND), only the N-terminal domains are degraded, resulting in the release of the bioactive C-terminal domains, which then forms an inflammasome. Thus, the N-terminal domains on NLRP1 function as a molecular tripwire to allow the sensing of pathogen-encoded proteases (and other effectors!). We named this mechanism Functional Degradation (see above Figure). Our publication describing this work can be found here.
We are actively pursuing a multitude of projects related to this idea, including the pathogen-specific activities that are sensed by human NLRP1. Our work on NLRP1 activation by viral proteases encoded by positive-sense RNA viruses can be found here.
We are broadly interested in the following research questions:
There are many models of infectious disease that have greatly informed our understanding of immunity and pathogenesis. However, human pathogens and human immunity and disease can be difficult to study because model organisms are often refractory to infection by pathogens that are highly adapted to the human host. Indeed, the absence of or variation in host proteins that serve as entry receptors for viruses is a classic example of species-specific complexity that effects host susceptibility to infection.
What is the source of this species-specific variation? In the MITCHELL LAB we are testing the hypothesis that much of this variation stems from host-pathogen evolutionary 'arms races' – the private history of pathogen-driven evolution uniquely shaping host immune repertoires in a lineage-specific manner. Our view is that beyond entry receptors, other 'mismatches' in host-pathogen interactions significantly effects the utility of human infectious disease models. For example, our finding that the NAIP–NLRC4 inflammasome protects mice from Shigella flexneri allowed us to develop inflammasome-deficient mice as the first physiologically relevant mouse model of human shigellosis.
We plan to leverage our understanding of species-specific host-pathogen interactions to generate mice, cells, and/or 3D ex vivo (i.e., organoid) cultures with ‘humanized’ genes using CRISPR-based editing strategies. We hope these models will better recapitulate human immunity, inflammation, and disease, as well as providing new tools to study microbial replication and pathogenesis.
